关于威尔逊病
症状
威尔逊病的临床特点:
肝
- 无症状性肝肿大
- 孤立性脾肿大
- 血清转氨酶持续升高(AST、ALT)
- 脂肪肝
- 急性肝炎
- 类似自身免疫性肝炎
- 肝硬化:补偿或失代偿
- 急性肝衰竭
神经系统
- 运动失调(震颤、不自主运动)
- 流口水,构音障碍
- 僵硬的肌张力障碍
- 假性球麻痹
- 自主神经紊乱
- 偏头痛
- 失眠症
- 癫痫发作
精神病学
- 抑郁
- 神经质行为
- 人格变化
- 精神病
其它系统
- 眼睛:K-F环,向日葵白内障
- 皮肤:新月形蓝印
- 肾脏异常:氨基酸尿症和肾结石
- 骨骼异常:骨质疏松和关节炎
- 心肌病、心律失常
- 胰腺炎
- 甲状旁腺功能减退症
- 月经不规律;不孕、反复流产
KF环
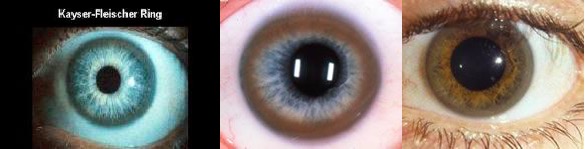
定义
Kayser Fleischer环(简称KF环):临床表现–棕黄色的环,角巩膜交界处可见。铜沉积于Descemet膜,延伸到骨小梁网。肝豆状核变性病征。(眼科术语词典第三版,Barbara Cassin, Sheila A.B. Solomon)
描述与定位
在角膜(Descemet的膜)的边缘中,存在金色或绿褐色的环形沉积。首先出现的是一个新月形,然后继续发展,最终变成了环状的。通常需要裂隙灯检查,方可发现早期形成的KF环。
患病率
约95%的神经系统症状的WD患者有K-F环,而约65%的肝脏症状的WD患者会出现一个K-F环。
铜螯合疗法可能导致角膜铜的褪色甚至消失。
诊断
Wilson病是怎么诊断的?
Wilson病的诊断是通过相对简单的测试进行的。这些测试可以在有症状的病人和没有疾病迹象的人中诊断出这种疾病。这些测试可包括:
- 对K-F环的裂隙灯检查
- 血清铜蓝蛋白试验
- 24小时尿铜检测
- 肝活检组织学、组织化学和铜定量
- 基因检测、兄弟姐妹的单倍型分析和突变分析
尽早诊断Wilson病是很重要的,因为严重的肝脏损伤可能会在出现任何疾病迹象之前发生。Wilson病患者可能会出现虚假的健康状况。
治疗
Wilson病是一种很容易治疗的疾病。通过适当的治疗,可以阻止疾病的发展,通常情况下症状能够改善。处理的目的是去除过剩的铜和防止其再积累。Wilson病的治疗是一个终生的过程。病人可能会变得越来越严重,所以立即治疗可能是至关重要的。治疗延误可能造成不可逆转的损害。
经批准用于治疗Wilson病的螯合治疗药物包括青霉胺(商品名Cuprimine®和Depen®)和盐酸曲恩汀(商品名Syprine®和Trientine®)。这两种药物都是通过铜的螯合或结合而起作用的,导致其尿排泄增加。
在美国,金属硫蛋白诱导剂被批准用于治疗Wilson病。锌通过阻断肠道中铜的吸收而起作用。这一作用既消耗积累的铜,又阻止其再积累。国外30多年的丰富经验证明了锌的有效性。锌疗法的一个主要优点是没有副作用。
重型肝炎或肝功能衰竭患者可能需要肝移植。接受Wilson病检查或治疗的病人应由Wilson病专家或专家与他们的主治医生联合诊治。完全停止治疗会导致死亡,最快的会在三个月内死亡。减少药物用量也会导致不必要的疾病发展。
按医嘱服药是对Wilson病治疗成功的至关重要因素。威尔逊疾病可以通过有效的,安全的药物治疗,从而达到正常的生活质量和寿命.前提是如果患者能够认真地按照医嘱终身服药。Wilson病患者会呈现不同的不依从原因,你的医生或许都见到过。
诊断时无症状的病人依从性尤其困难,因为这些患者往往看不到不好好服药会造成很严重的后果。
Gluzin
Gluzin™是药品级锌盐,其活性成分是葡萄糖酸锌,提供了良好的锌矿物质。锌盐有很多种,但葡萄糖酸锌比其他锌盐更易溶解,而且对胃刺激小。
现在可用:Gluzin ™易吞的葡萄糖酸锌胶囊(25毫克和50毫克),有助于:
- 阻断多余铜在体内的吸收
- 保持健康锌水平。
- 支持健康的免疫系统
- 平衡人体必需微量元素的相互作用
- 支持健康的DNA合成和代谢
Gluzin™已制定与最低限度的副料,以帮助减少过敏反应。有关Gluzin的更多信息™,请登录www.extremeV.com或电邮至info@extremev.com。
遗传
威尔逊病如何遗传?
威尔逊病是一种常染色体隐性遗传性疾病,这意味着它在男性和女性中平等地发生。父母都携带一个遗传突变(基因异常改变),双方把各自的致病突变传给孩子,孩子就会得威尔逊病。此病在所有已知种族和民族中的发病率至少为3万分之一。
23种不同的人类染色体中,导致威尔逊病的基因位于13号染色体上。该基因称为ATP7B,其含有合成铜转运蛋白必需的遗传信息。铜转运蛋白在铜蓝蛋白结合铜和把多余的铜排出肝脏方面发挥关键作用。
该基因突变导致铜转运蛋白异常,排铜能力部分或全部缺失。到目前为止,已知300多种ATB7B基因。这种多余的铜积聚在肝脏和其他器官中。
多数患者无家族史。只有一个异常基因的人被称为携带者。(杂合子)可能具有轻度的(但医学上无关紧要的)铜代谢异常。携带者不会发病,不用接受治疗。威尔逊病患者(纯合子)会发病,必须接受终身治疗,否则最终会发展为严重的致命疾病。
遗传威尔逊病的可能性?
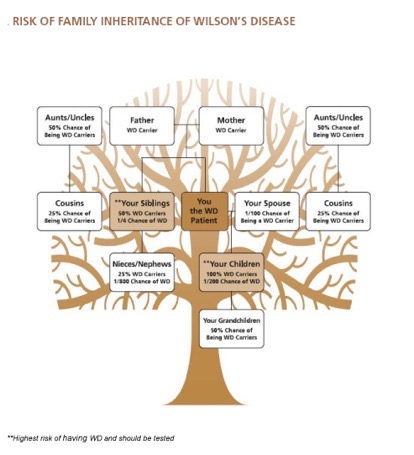
一般人群中威尔逊病致病基因的携带者为一百分之一。携带者有一个正常和一个异常基因。威尔逊病患者的所有孩子至少都携带了一个威尔逊病致病基因。携带者的孩子携带致病基因的概率为50%。该图是家庭成员的基本风险简介。遗传咨询师可以提供更详细的家族关系谱系。
家庭筛选
威尔逊病患者的所有兄弟姐妹和孩子都应接受威尔逊病检测。患者亲属,如果已经出现症状,或做了检查表明有相关的肝脏或神经系统疾病,也应进行威尔逊病检测。
生化检测
儿童患者 —— 如果无症状,从2岁开始,5年内重复一次,除非有进一步排查的理由。
患者兄弟姐妹
- 身体检查和任何肝脏或神经系统症状的病史
- 肝功能检测:ALT,AST,白蛋白,胆红素。
- 血浆铜蓝蛋白和血清铜。
- 24小时尿铜
- 裂缝灯Kayser-Fleischer(K-F)环检查。
- 如果没有K-F环,肝功能无异常,铜蓝蛋白不低:行肝活检
关于分子遗传测试的信息
威尔逊病患者的所有兄弟姐妹和孩子都应接受威尔逊病检测。其他已经出现症状或实验室检查,表明肝脏或神经系统疾病的亲属也应进行Wilson病检测。到目前为止,已经鉴定了ATP7B基因的300多个不同突变。
测试方法
连锁分析(单倍型分析)
分子遗传学检测来鉴定一组紧密连锁的DNA片段(一个标记或一组标记),将家族成员的标记与受影响的患者的标记进行比较。
用于:筛选鉴定患者的兄弟姐妹
基因测序(整个ATP7B基因的突变筛选)
分析整个ATP7B基因以检测和鉴定引起疾病的突变。确诊威尔逊病患者首先需要进行检测,如果两个突变都被鉴定出来,那么其他家族成员也需要进行测试。基因测序在大多数情况下都能识别出这两种突变,但并不是所有的威尔逊病病例。
基因测序可用于:疑似患者的诊断,排查尚未有症状的患者亲属是否同患此病或是否携带,或对确认的携带者进行产前检测。
目标突变分析
对已知特定突变的ATP7B基因的特定位置进行分析。
适用于:已知常见突变的患者的特定人群;对有两个确定突变的患者的兄弟姐妹进行筛选。基因测试最好通过一个遗传顾问进行,他们可以仔细讨论测试的最佳方法,以及每种方法的好处、限制和影响。你的医生应该能够指导你找到一个合格的遗传顾问和基因检测设施。
威尔逊疾病协会
威尔逊疾病是一种以青少年为主的常染色体隐性遗传性疾病,是先天性铜代谢障碍性疾病。临床上以肝损害、锥体外系症状与角膜色素环等为主要表现,伴有血浆铜蓝蛋白缺少和氨基酸尿症。